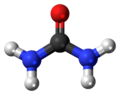
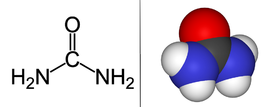
Urea, also known as carbamide, is an organic compound with chemical formula CO(NH2)2. This amide has two –NH2 groups joined by a carbonyl (C=O) functional group.
Urea serves an important role in the metabolism of nitrogen-containing compounds by animals and is the main nitrogen-containing substance in the urine of mammals. It is a colorless, odorless solid, highly soluble in water, and practically non-toxic (LD50 is 15 g/kg for rats).[5] Dissolved in water, it is neither acidic nor alkaline. The body uses it in many processes, most notably nitrogen excretion. The liver forms it by combining two ammonia molecules (NH3) with a carbon dioxide (CO2) molecule in the urea cycle. Urea is widely used in fertilizers as a source of nitrogen and is an important raw material for the chemical industry.
Friedrich Wöhler's discovery in 1828 that urea can be produced from non-biological starting materials was an important conceptual milestone in chemistry. It showed for the first time that a substance previously known only as a byproduct of life could be synthesized in the laboratory without biological starting materials thereby contradicting the widely held doctrine of vitalism.
Uses
Agriculture
A process plant in Bangladesh that commercially produces urea as fertilizer by using methane as the main raw material.
More than 90% of world industrial production of urea is destined for use as a nitrogen-release fertilizer.[6] Urea has the highest nitrogen content of all solid nitrogenous fertilizers in common use. Therefore, it has the lowest transportation costs per unit of nitrogen nutrient.
Many soil bacteria possess the enzyme urease, which catalyzes conversion of urea to ammonia (NH3) or ammonium ion (NH4+) and bicarbonate ion (HCO3−). Thus urea fertilizers rapidly transform to the ammonium form in soils. Among the soil bacteria known to carry urease, some ammonia-oxidizing bacteria (AOB), such as species of Nitrosomonas, can also assimilate the carbon dioxide the reaction releases to make biomass via the Calvin cycle, and harvest energy by oxidizing ammonia (the other product of urease) to nitrite, a process termed nitrification.[7] Nitrite-oxidizing bacteria, especially Nitrobacter, oxidize nitrite to nitrate, which is extremely mobile in soils because of its negative charge and is a major cause of water pollution from agriculture. Ammonium and nitrate are readily absorbed by plants, and are the dominant sources of nitrogen for plant growth. Urea is also used in many multi-component solid fertilizer formulations. Urea is highly soluble in water and is therefore also very suitable for use in fertilizer solutions (in combination with ammonium nitrate: UAN), e.g., in 'foliar feed' fertilizers. For fertilizer use, granules are preferred over prills because of their narrower particle size distribution, which is an advantage for mechanical application.
The most common impurity of synthetic urea is biuret, which impairs plant growth.
Urea is usually spread at rates of between 40 and 300 kg/ha but rates vary. Smaller applications incur lower losses due to leaching. During summer, urea is often spread just before or during rain to minimize losses from volatilization (a process wherein nitrogen is lost to the atmosphere as ammonia gas).
Because of the high nitrogen concentration in urea, it is very important to achieve an even spread. The application equipment must be correctly calibrated and properly used. Drilling must not occur on contact with or close to seed, due to the risk of germination damage. Urea dissolves in water for application as a spray or through irrigation systems.
In grain and cotton crops, urea is often applied at the time of the last cultivation before planting. In high rainfall areas and on sandy soils (where nitrogen can be lost through leaching) and where good in-season rainfall is expected, urea can be side- or top-dressed during the growing season. Top-dressing is also popular on pasture and forage crops. In cultivating sugarcane, urea is side-dressed after planting, and applied to each ratooncrop.
In irrigated crops, urea can be applied dry to the soil, or dissolved and applied through the irrigation water. Urea dissolves in its own weight in water, but becomes increasingly difficult to dissolve as the concentration increases. Dissolving urea in water is endothermic—the solution temperature falls when urea dissolves.
As a practical guide, when preparing urea solutions for fertigation (injection into irrigation lines), dissolve no more than 3 g urea per 1 L water.
In foliar sprays, urea concentrations of between 0.5% and 2.0% are often used in horticultural crops. Low-biuret grades of urea are often indicated.
Urea absorbs moisture from the atmosphere and therefore is typically stored either in closed or sealed bags on pallets or, if stored in bulk, under cover with a tarpaulin. As with most solid fertilizers, storage in a cool, dry, well-ventilated area is recommended.
Overdose or placing urea near seed is harmful.[8]
Chemical industry
Urea is a raw material for the manufacture of two main classes of materials: urea-formaldehyde resins and urea-melamine-formaldehyde used in marine plywood.
Explosives
Urea can be used to make urea nitrate, a high explosive that is used industrially and as part of some improvised explosive devices. It is a stabilizer in nitrocellulose explosives.
Automobile systems
Urea is used in SNCR and SCR reactions to reduce the NOx pollutants in exhaust gases from combustion from Diesel, dual fuel, and lean-burn natural gas engines. The BlueTec system, for example, injects a water-based urea solution into the exhaust system. The ammonia produced by the hydrolysis of the urea reacts with the nitrogen oxide emissions and is converted into nitrogen and water within the catalytic converter. Trucks and cars using these catalytic converters need to carry a supply of diesel exhaust fluid, a solution of urea in water.
Laboratory uses
Urea in concentrations up to 10 M is a powerful protein denaturant as it disrupts the noncovalent bonds in the proteins. This property can be exploited to increase the solubility of some proteins. A mixture of urea and choline chloride is used as a deep eutectic solvent, a type of ionic liquid.
Urea can in principle serve as a hydrogen source for subsequent power generation in fuel cells. Urea present in urine/wastewater can be used directly (though bacteria normally quickly degrade urea.) Producing hydrogen by electrolysis of urea solution occurs at a lower voltage (0.37 V) and thus consumes less energy than the electrolysis of water (1.2 V).[9]
Urea in concentrations up to 8 M can be used to make fixed brain tissue transparent to visible light while still preserving fluorescent signals from labeled cells. This allows for much deeper imaging of neuronal processes than previously obtainable using conventional one photon or two photon confocal microscopes.[10]
Medical use
Urea-containing creams are used as topical dermatological products to promote rehydration of the skin. Urea 40% is indicated for psoriasis, xerosis, onychomycosis, ichthyosis, eczema, keratosis, keratoderma, corns, and calluses. If covered by an occlusive dressing, 40% urea preparations may also be used for nonsurgical debridement of nails. Urea 40% "dissolves the intercellular matrix"[11] of the nail plate. Only diseased or dystrophic nails are removed, as there is no effect on healthy portions of the nail.[citation needed] This drug is also used as an earwax removal aid.[citation needed]
Urea has also been studied as a diuretic. It was first used Dr. W. Friedrich in 1892.[12] In a 2010 study of ICU patients, urea was used to treat euvolemic hyponatremia and was found safe, inexpensive, and simple.[13]
Like saline, urea injection has previously been used to perform abortion.[14]
The blood urea nitrogen (BUN) test is a measure of the amount of nitrogen in the blood that comes from urea. It is used as a marker of renal function, though it is inferior to other markers such as creatinine because blood urea levels are influenced by other factors such as diet and dehydration.[15]
Urea labeled with carbon-14 or carbon-13 is used in the urea breath test, which is used to detect the presence of the bacterium Helicobacter pylori (H. pylori) in the stomach and duodenum of humans, associated with peptic ulcers. The test detects the characteristic enzyme urease, produced by H. pylori, by a reaction that produces ammonia from urea. This increases the pH (reduces the acidity) of the stomach environment around the bacteria. Similar bacteria species to H. pylori can be identified by the same test in animals such as apes, dogs, and cats (including big cats).
Miscellaneous uses
• A component of animal feed, providing a relatively cheap source of nitrogen to promote growth
• A non-corroding alternative to rock salt for road de-icing.[16] It is often the main ingredient of pet friendly salt substitutes although it is less effective than traditional rock salt or calcium chloride.[17]
• A main ingredient in hair removers such as Nair and Veet
• A browning agent in factory-produced pretzels
• An ingredient in some skin cream,[18] moisturizers, hair conditioners, and shampoos
• A cloud seeding agent, along with other salts (citation needed)
• A flame-proofing agent, commonly used in dry chemical fire extinguisher charges such as the urea-potassium bicarbonate mixture
• An ingredient in many tooth whitening products
• An ingredient in dish soap
• Along with diammonium phosphate, as a yeast nutrient, for fermentation of sugars into ethanol
• A nutrient used by plankton in ocean nourishment experiments for geoengineering purposes
• As an additive to extend the working temperature and open time of hide glue
• As a solubility-enhancing and moisture-retaining additive to dye baths for textile dyeing or printing
• As an optical parametric oscillator in nonlinear optics
Adverse effects
Urea can be irritating to skin, eyes, and the respiratory tract. Repeated or prolonged contact with urea in fertilizer form on the skin may cause dermatitis.[citation needed]
High concentrations in the blood can be damaging. Ingestion of low concentrations of urea, such as are found in typical human urine, are not dangerous with additional water ingestion within a reasonable time-frame. Many animals (e.g., dogs) have a much more concentrated urine and it contains a higher urea amount than normal human urine; this can prove dangerous as a source of liquids for consumption in a life-threatening situation (such as in a desert).
Urea can cause algal blooms to produce toxins, and its presence in the runoff from fertilized land may play a role in the increase of toxic blooms.[21]
The substance decomposes on heating above melting point, producing toxic gases, and reacts violently with strong oxidants, nitrites, inorganic chlorides, chlorites and perchlorates, causing fire and explosion.
Physiology
Amino acids from ingested food that are not used for the synthesis of proteins and other biological substances — or produced from catabolism of muscle protein — are oxidized by the body as an alternative source of energy, yielding urea and carbon dioxide.[23] The oxidation pathway starts with the removal of the amino group by a transaminase; the amino group is then fed into the urea cycle. The first step in the conversion of amino acids from protein into metabolic waste in the liver is removal of the alpha-amino nitrogen, which results in ammonia. Because ammonia is toxic, it is excreted immediately by fish, converted into uric acid by birds, and converted into urea by mammals.[24]
Ammonia (NH3) is a common byproduct of the metabolism of nitrogenous compounds. Ammonia is smaller, more volatile and more mobile than urea. If allowed to accumulate, ammonia would raise the pH in cells to toxic levels. Therefore, many organisms convert ammonia to urea, even though this synthesis has a net energy cost. Being practically neutral and highly soluble in water, urea is a safe vehicle for the body to transport and excrete excess nitrogen.
Urea is synthesized in the body of many organisms as part of the urea cycle, either from the oxidation of amino acids or from ammonia. In this cycle, amino groups donated by ammonia and L-aspartate are converted to urea, while L-ornithine, citrulline, L-argininosuccinate, and L-arginine act as intermediates. Urea production occurs in the liver and is regulated by N-acetylglutamate. Urea is then dissolved into the blood (in the reference range of 2.5 to 6.7 mmol/liter) and further transported and excreted by the kidney as a component of urine. In addition, a small amount of urea is excreted (along with sodium chloride and water) in sweat.
In water, the amine groups undergo slow displacement by water molecules, producing ammonia, ammonium ion, and bicarbonate ion. For this reason, old, stale urine has a stronger odor than fresh urine.
Humans
The cycling of and excretion of urea by the kidneys is a vital part of mammalian metabolism. Besides its role as carrier of waste nitrogen, urea also plays a role in the countercurrent exchange systemof the nephrons, that allows for re-absorption of water and critical ions from the excreted urine. Urea is reabsorbed in the inner medullary collecting ducts of the nephrons,[25] thus raising the osmolarity in the medullary interstitium surrounding the thin descending limb of the loop of Henle, which makes the water reabsorb.
By action of the urea transporter 2, some of this reabsorbed urea eventually flows back into the thin descending limb of the tubule,[26] through the collecting ducts, and into the excreted urine. The body uses this mechanism, which is controlled by the antidiuretic hormone, to create hyperosmotic urine—i.e., urine with a higher concentration of dissolved substances than the blood plasma. This mechanism is important to prevent the loss of water, maintain blood pressure, and maintain a suitable concentration of sodium ions in the blood plasma.
The equivalent nitrogen content (in gram) of urea (in mmol) can be estimated by the conversion factor 0.028 g/mmol.[27] Furthermore, 1 gram of nitrogen is roughly equivalent to 6.25 grams of protein, and 1 gram of protein is roughly equivalent to 5 grams of muscle tissue. In situations such as muscle wasting, 1 mmol of excessive urea in the urine (as measured by urine volume in litres multiplied by urea concentration in mmol/l) roughly corresponds to a muscle loss of 0.67 gram.
Other species
In aquatic organisms the most common form of nitrogen waste is ammonia, whereas land-dwelling organisms convert the toxic ammonia to either urea or uric acid. Urea is found in the urine of mammals and amphibians, as well as some fish. Birds and saurian reptiles have a different form of nitrogen metabolism that requires less water, and leads to nitrogen excretion in the form of uric acid. It is noteworthy that tadpoles excrete ammonia but shift to urea production during metamorphosis. Despite the generalization above, the urea pathway has been documented not only in mammals and amphibians but in many other organisms as well, including birds, invertebrates, insects, plants, yeast, fungi, and even microorganisms.[citation needed]
Analysis
Urea is readily quantified by a number of different methods, such as the diacetyl monoxime colorimetric method, and the Berthelot reaction (after initial conversion of urea to ammonia via urease). These methods are amenable to high throughput instrumentation, such as automated flow injection analyzers[28] and 96-well micro-plate spectrophotometers.[29]
Related compounds
Main article: ureas
Ureas describes a class of chemical compounds that share the same functional group, a carbonyl group attached to two organic amine residues: RR'N—CO—NRR'. Examples include carbamide peroxide, allantoin, and hydantoin. Ureas are closely related to biurets and related in structure to amides, carbamates, carbodiimides, and thiocarbamides.
History
Urea was first discovered in urine in 1727 by the Dutch scientist Herman Boerhaave,[30] although this discovery is often attributed to the French chemist Hilaire Rouelle.[31]
Boerhaave used the following steps to isolate urea:[32][33]
1. Boiled off water, resulting in a substance similar to fresh cream
2. Used filter paper to squeeze out remaining liquid
3. Waited a year for solid to form under an oily liquid
4. Removed the oily liquid
5. Dissolved the solid in water
6. Used recrystallization to tease out the urea
In 1828, the German chemist Friedrich Wöhler obtained urea artificially by treating silver cyanate with ammonium chloride.[34][35][36]
AgNCO + NH4Cl → (NH2)2CO + AgCl
This was the first time an organic compound was artificially synthesized from inorganic starting materials, without the involvement of living organisms. The results of this experiment implicitly discredited vitalism — the theory that the chemicals of living organisms are fundamentally different from those of inanimate matter. This insight was important for the development of organic chemistry. His discovery prompted Wöhler to write triumphantly to Berzelius: "I must tell you that I can make urea without the use of kidneys, either man or dog. Ammonium cyanate is urea." In fact, this was incorrect. These are two different chemicals, which are in chemical equilibrium heavily favoring urea under standard conditions.[37] Regardless, with his discovery, Wöhler secured a place among the pioneers of organic chemistry.
Production
Urea is produced on an industrial scale: In 2012, worldwide production capacity was approximately 184 million tonnes.
Industrial methods
For use in industry, urea is produced from synthetic ammonia and carbon dioxide. As large quantities of carbon dioxide are produced during the ammonia manufacturing process as a byproduct from hydrocarbons (predominantly natural gas, less often petroleum derivatives), or occasionally from coal, urea production plants are almost always located adjacent to the site where the ammonia is manufactured. Although natural gas is both the most economical and the most widely available ammonia plant feedstock, plants using it do not produce quite as much carbon dioxide from the process as is needed to convert their entire ammonia output into urea. In recent years new technologies such as the KM-CDR process have been developed to recover supplementary carbon dioxide from the combustion exhaust gases produced in the fired reforming furnace of the ammonia synthesis gas plant, allowing operators of stand-alone nitrogen fertilizer complexes to avoid the need to handle and market ammonia as a separate product and also to reduce their
greenhouse gas emissions to the atmosphere.
Synthesis
The basic process, developed in 1922, is also called the Bosch–Meiser urea process after its discoverers. Various commercial urea processes are characterized by the conditions under which urea forms and the way that unconverted reactants are further processed. The process consists of two main equilibrium reactions, with incomplete conversion of the reactants. The first is carbamate formation: the fast exothermic reaction of liquid ammonia with gaseous carbon dioxide (CO2) at high temperature and pressure to form ammonium carbamate (H2N-COONH4)
2 NH3 + CO2 ⇌ H2N-COONH4 (ΔH= -117kJ/mol at 110 atm and 160°C) [42]
The second is urea conversion: the slower endothermic decomposition of ammonium carbamate into urea and water:
H2N-COONH4 ⇌ (NH2)2CO + H2O (ΔH= +15.5 kJ/mol at 160-180°C) [42]
The overall conversion of NH3 and CO2 to urea is exothermic,[6] the reaction heat from the first reaction driving the second. Like all chemical equilibria, these reactions behave according to Le Chatelier's principle, and the conditions that most favour carbamate formation have an unfavourable effect on the urea conversion equilibrium. The process conditions are, therefore, a compromise: the ill-effect on the first reaction of the high temperature (around 190 °C) needed for the second is compensated for by conducting the process under high pressure (140–175 bar), which favours the first reaction. Although it is necessary to compress gaseous carbon dioxide to this pressure, the ammonia is available from the ammonia plant in liquid form, which can be pumped into the system much more economically. To allow the slow urea formation reaction time to reach equilibrium a large reaction space is needed, so the synthesis reactor in a large urea plant tends to be a massive pressure vessel.
Because the urea conversion is incomplete, the product must be separated from unchanged ammonium carbamate. In early "straight-through" urea plants this was done by letting down the system pressure to atmospheric to let the carbamate decompose back to ammonia and carbon dioxide. Originally, because it was not economic to recompress the ammonia and carbon dioxide for recycle, the ammonia at least would be used for the manufacture of other products, for example ammonium nitrate or sulfate. (The carbon dioxide was usually wasted.) Later process schemes made recycling unused ammonia and carbon dioxide practical. This was accomplished by depressurizing the reaction solution in stages (first to 18–25 bar and then to 2–5 bar) and passing it at each stage through a steam-heated carbamate decomposer, then recombining the resultant carbon dioxide and ammonia in a falling-film carbamate condenser and pumping the carbamate solution into the previous stage.
The stripping concept
The "total recycle" concept has two main disadvantages. The first is the complexity of the flow scheme and, consequently, the amount of process equipment needed. The second is the amount of water recycled in the carbamate solution, which has an adverse effect on the equilibrium in the urea conversion reaction and thus on overall plant efficiency. The stripping concept, developed in the early 1960s by Stamicarbon in The Netherlands, addressed both problems. It also improved heat recovery and reuse in the process.
The position of the equilibrium in the carbamate formation/decomposition depends on the product of the partial pressures of the reactants. In the total recycle processes, carbamate decomposition is promoted by reducing the overall pressure, which reduces the partial pressure of both ammonia and carbon dioxide. It is possible, however, to achieve a similar effect without lowering the overall pressure—by suppressing the partial pressure of just one of the reactants. Instead of feeding carbon dioxide gas directly to the reactor with the ammonia, as in the total recycle process, the stripping process first routes the carbon dioxide through a stripper (a carbamate decomposer that operates under full system pressure and is configured to provide maximum gas-liquid contact). This flushes out free ammonia, reducing its partial pressure over the liquid surface and carrying it directly to a carbamate condenser (also under full system pressure). From there, reconstituted ammonium carbamate liquor passes directly to the reactor. That eliminates the medium-pressure stage of the total recycle process altogether.
The stripping concept was such a major advance that competitors such as Snamprogetti—now Saipem—(Italy), the former Montedison (Italy), Toyo Engineering Corporation (Japan), and Urea Casale(Switzerland) all developed versions of it. Today, effectively all new urea plants use the principle, and many total recycle urea plants have converted to a stripping process. No one has proposed a radical alternative to the approach. The main thrust of technological development today, in response to industry demands for ever larger individual plants, is directed at re-configuring and re-orientating major items in the plant to reduce size and overall height of the plant, and at meeting challenging environmental performance targets.[43][44]
Side reactions
It is fortunate that the urea conversion reaction is slow. If it were not it would go into reverse in the stripper. As it is, succeeding stages of the process must be designed to minimize residence times, at least until the temperature reduces to the point where the reversion reaction is very slow.
Two reactions produce impurities. Biuret is formed when two molecules of urea combine with the loss of a molecule of ammonia.
2 NH2CONH2 → H2NCONHCONH2 + NH3
Normally this reaction is suppressed in the synthesis reactor by maintaining an excess of ammonia, but after the stripper, it occurs until the temperature is reduced. Biuret is undesirable in fertilizer urea because it is toxic to crop plants, although to what extent depends on the nature of the crop and the method of application of the urea.[45] (Biuret is actually welcome in urea when is used as a cattle feed supplement).
Isocyanic acid results from the thermal decomposition of ammonium cyanate, which is in chemical equilibrium with urea:
NH2CONH2 → NH4NCO → HNCO + NH3
This reaction is at its worst when the urea solution is heated at low pressure, which happens when the solution is concentrated for prilling or granulation (see below). The reaction products mostly volatilize into the overhead vapours, and recombine when these condense to form urea again, which contaminates the process condensate.
Corrosion
Ammonium carbamate solutions are notoriously corrosive to metallic construction materials, even more resistant forms of stainless steel—especially in the hottest parts of the plant such as the stripper. Historically corrosion has been minimized (although not eliminated) by continuous injection of a small amount of oxygen (as air) into the plant to establish and maintain a passive oxide layer on exposed stainless steel surfaces. Because the carbon dioxide feed is recovered from ammonia synthesis gas, it contains traces of hydrogen that can mingle with passivation air to form an explosive mixture if allowed to accumulate.
In the mid 1990s two duplex (ferritic-austenitic) stainless steels were introduced (DP28W, jointly developed by Toyo Engineering and Sumitomo Metals Industries and Safurex, jointly developed by Stamicarbon and Sandvik Materials Technology (Sweden).) These let manufactures drastically reduce the amount of passivation oxygen. In theory, they could operate with no oxygen.
Saipem now uses either zirconium stripper tubes, or bimetallic tubes with a titanium body (cheaper but less erosion-resistant) and a metallurgically bonded internal zirconium lining. These tubes are fabricated by ATI Wah Chang (USA) using its Omegabond technique.
Finishing
Urea can be produced as prills, granules, pellets, crystals, and solutions.
Solid forms
For its main use as a fertilizer urea is mostly marketed in solid form, either as prills or granules. The advantage of prills is that, in general, they can be produced more cheaply than granules and that the technique was firmly established in industrial practice long before a satisfactory urea granulation process was commercialized. However, on account of the limited size of particles that can be produced with the desired degree of sphericity and their low crushing and impact strength, the performance of prills during bulk storage, handling and use is generally (with some exceptions[50]) considered inferior to that of granules.
High-quality compound fertilizers containing nitrogen co-granulated with other components such as phosphates have been produced routinely since the beginnings of the modern fertilizer industry, but on account of the low melting point and hygroscopic nature of urea it took courage to apply the same kind of technology to granulate urea on its own. But at the end of the 1970s three companies began to develop fluidized-bed granulation. The first in the field was Nederlandse Stikstof Maatschappij, which later became part of Hydro Agri (now Yara International). Yara eventually sold this technology to Uhde GmbH, whose Uhde Fertilizer Technology (UFT) subsidiary now markets it. Around the same time Toyo Engineering Corporation developed its spouted-bed process, comprising a fluidized bed deliberately agitated to produce turbulent ebullation. Stamicarbon also undertook development work on its own fluidized-bed granulation system, using film sprays rather than atomizing sprays to introduce the urea melt, but shelved it until the 1990s, when there was for a time considerable doubt about the commercial future of the Hydro (UFT) process. As a result, the Stamicarbon technology is now commercialized and highly successful. More recently, Urea Casale introduced a different fluidized-bed granulation system: the urea is sprayed in laterally from the side walls of the granulator instead of from the bottom. This organizes the bed into two cylindrical masses contrarotating on parallel longitudinal axes. The raw product is uniform enough not to require screens.
Surprisingly, perhaps, considering the product particles are not spherical, pastillation using a Rotoform steel-belt pastillator is gaining ground as a urea particle-forming process as a result of development work by Stamicarbon in collaboration with Sandvik Process Systems (Germany). Single-machine capacity is limited to 175 t/d, but the machines are simple and need little maintenance, specific power consumption is much lower than for granulation, and the product is very uniform. The robustness of the product appears to make up for its non-spherical shape.
UAN solutions
In admixture, the combined solubility of ammonium nitrate and urea is so much higher than that of either component alone that it is possible to obtain a stable solution (known as UAN) with a total nitrogen content (32%) approaching that of solid ammonium nitrate (33.5%), though not, of course, that of urea itself (46%). Given the ongoing safety and security concerns surrounding fertilizer-grade solid ammonium nitrate, UAN provides a considerably safer alternative without entirely sacrificing the agronomic properties that make ammonium nitrate more attractive than urea as a fertilizer in areas with short growing seasons. It is also more convenient to store and handle than a solid product and easier to apply accurately to the land by mechanical means.
Laboratory preparation
Ureas in the more general sense can be accessed in the laboratory by reaction of phosgene with primary or secondary amines, proceeding through an isocyanate intermediate. Non-symmetric ureas can be accessed by reaction of primary or secondary amines with an isocyanate.
Also, urea is produced when phosgene reacts with ammonia:
COCl2 + 4 NH3 → (NH2)2CO + 2 NH4Cl
Urea is byproduct of converting alkyl halides to thiols via a S-alkylation of thiourea. Such reactions proceed via the intermediacy of isothiouronium salts:
RX + CS(NH2)2 → RSCX(NH2)2X
RSCX(NH2)2X + MOH → RSH + (NH2)2CO + MX
In this reaction R is alkyl group, X is halogen and M is alkali metal.
Historical process
Urea was first noticed by Hermann Boerhaave in the early 18th century from evaporates of urine. In 1773, Hilaire Rouelle obtained crystals containing urea from human urine by evaporating it and treating it with alcohol in successive filtrations.[60] This method was aided by Carl Wilhelm Scheele's discovery that urine treated by concentrated nitric acid precipitated crystals. Antoine François, comte de Fourcroy and Louis Nicolas Vauquelin discovered in 1799 that the nitrated crystals were identical to Rouelle's substance and invented the term "urea. Berzelius made further improvements to its purification and finally William Prout, in 1817, succeeded in obtaining and determining the chemical composition of the pure substance. In the evolved procedure, urea was precipitated as urea nitrate by adding strong nitric acid to urine. To purify the resulting crystals, they were dissolved in boiling water with charcoal and filtered. After cooling, pure crystals of urea nitrate form. To reconstitute the urea from the nitrate, the crystals are dissolved in warm water, and barium carbonate added. The water is then evaporated and anhydrous alcohol added to extract the urea. This solution is drained off and evaporated, leaving pure urea.
Chemical properties
Molecular and crystal structure
The urea molecule is planar in the crystal structure, but the geometry around the nitrogens is pyramidal in the gas-phase minimum-energy structure. In solid urea, the oxygen center is engaged in two N-H-O hydrogen bonds. The resulting dense and energetically favourable hydrogen-bond network is probably established at the cost of efficient molecular packing: The structure is quite open, the ribbons forming tunnels with square cross-section. The carbon in urea is described as sp2 hybridized, the C-N bonds have significant double bond character, and the carbonyl oxygen is basic compared to, say, formaldehyde. Urea's high aqueous solubility reflects its ability to engage in extensive hydrogen bonding with water.
By virtue of its tendency to form porous frameworks, urea has the ability to trap many organic compounds. In these so-called clathrates, the organic "guest" molecules are held in channels formed by interpenetrating helices composed of hydrogen-bonded urea molecules. This behaviour can be used to separate mixtures, e.g., in the production of aviation fuel and lubricating oils, and in the separation of hydrocarbons.
As the helices are interconnected, all helices in a crystal must have the same molecular handedness. This is determined when the crystal is nucleated and can thus be forced by seeding. The resulting crystals have been used to separate racemic mixtures.
Reactions
Urea dissolved in water is in equilibrium with the isomeric ammonium cyanate. The resulting activity of the isocyanic acid ions do result in carbamylation (formation of long-chain carbamides, liberating ammonia molecule as byproduct) of proteins if proteins are present in the solution too. The carbamylation reaction may occur at elevated temperatures even without catalysts. At room temperature, water solutions of urea are prone to same decomposition reaction in the presence of urease. The isomerization of urea in solution at room temperature without catalysts is a slow process (taking days to reach equilibrium), and freshly prepared, unheated solutions had negligible carbamylation rates.
Urea reacts with alcohols to form urethanes.
Urea reacts with malonic esters to make barbituric acids.
Etymology and pronunciation
Urea is New Latin from French urée, from Ancient Greek οὖρον ouron, "urine".
|
Names |
|
|
Pronunciation |
urea /jʊəˈriːə/, carbamide /ˈkɑːrbəmaɪd/ |
|
Preferred IUPAC name Urea[1] |
|
|
Systematic IUPAC name Carbonyl diamide[1] |
|
|
Other names Carbamide |
|
|
Identifiers |
|
|
CAS Number |
· 57-13-6 |
|
3D model (JSmol) |
· Interactive image |
|
Beilstein Reference |
635724 |
|
ChEBI |
· CHEBI:16199 |
|
ChEMBL |
· ChEMBL985 |
|
ChemSpider |
· 1143 |
|
DrugBank |
· DB03904 |
|
ECHA InfoCard |
100.000.286 |
|
E number |
E927b (glazing agents, ...) |
|
Gmelin Reference |
1378 |
|
IUPHAR/BPS |
· 4539 |
|
KEGG |
· D00023 |
|
PubChem CID |
· 1176 |
|
RTECS number |
YR6250000 |
|
UNII |
· 8W8T17847W |
|
InChI[show] |
|
|
SMILES[show] |
|
|
Properties |
|
|
Chemical formula |
CH4N2O |
|
Molar mass |
60.056 g·mol−1 |
|
Appearance |
White solid |
|
Density |
1.32 g/cm3 |
|
Melting point |
133 to 135 °C (271 to 275 °F; 406 to 408 K) |
|
Solubility in water |
1079 g/L (20 °C) |
|
Solubility |
500 g/L glycerol[2] 50g/L ethanol |
|
Basicity (pKb) |
13.9[4] |
|
Magnetic susceptibility (χ) |
-33.4·10−6 cm3/mol |
|
Structure |
|
|
Dipole moment |
4.56 D |
|
ThermochemistryCRC Handbook |
|
|
Std enthalpy of |
-79.634 kcal/mol |
|
Gibbs free energy(ΔfG˚) |
-47.12 kcal/mol |
|
Pharmacology |
|
|
ATC code |
B05BC02 (WHO) D02AE01(WHO) |
|
Hazards |
|
|
Safety data sheet |
JT Baker |
|
GHS pictograms |
|
|
NFPA 704 |
1 1 0 |
|
Flash point |
Non-flammable |
|
Lethal dose or concentration (LD, LC): |
|
|
LD50 (median dose) |
8500 mg/kg (oral, rat) |
|
Related compounds |
|
|
Related ureas |
Thiourea |
|
Related compounds |
Carbamide peroxide |
|
Except where otherwise noted, data are given for materials in their standard state (at 25 °C [77 °F], 100 kPa). |
|
|
verify (what is ?) |
|
|
Infobox references |
|